
涂料技术
UV固化涂料用新型高效光引发剂的研制开发
[据《现代涂料与涂装》报道] 作者:吴光辉 ( 青岛帝科精细化学有限公司,山东青岛 )
摘要:为了进一步拓展紫外光固化涂料的用途,解决黄变问题 ,设计了紫外光敏感性和引发活性非常高的新型光引发剂—— 丙烯酸甲酯基三乙酰基甲烷,并用 1H — NMR 13C — NMR 、 GC — MS 进行了表征。
关键词:光引发剂;乙酰丙酮;丙烯酸甲酯基三乙酰基甲烷;紫外光固化涂料
紫外光固化粉末涂料 ( 简称 uV 固化粉末涂料 ) 是 一 项将传统粉末涂料和 uV 固化技术相结合的新技 术,兼备两项技术的优点,特别适合于需要环境保护、 低温涂装、快速固化、高效率生产的场合。目前 uv 固化粉末涂料已解决了常规热固性粉末涂料不能用于热敏材料的问题。 uV 固化粉末涂料属“ 4E ”技术,而且具有非常大的市场潜力,因此,对它的研究出现了日 新月异的发展。
uV 固化粉末涂料组分有:光引发剂、主体树脂、固化剂和各种助剂。这种涂料以高能量的紫外光作为固化能源,由涂料中的光引发剂吸收紫外光,分子从基态跃迁到活泼的激发单线态,还可继续经系间窜越,跃迁至激发三线态。在其激发单线态,也可能是在激发三线态经历单分子或双分子化学作用后,产生活性碎片,这些碎片可以是自由基或离子,从而引发体系中光敏树脂和活性稀释剂中的不饱和键聚合,使涂膜完成固化。因此光引发剂是配方中必不可少的组分,光引发剂的物理和化学性质对光引发、光聚合反应过程的控制非常重要。根据光引发机理分类, uV 固化粉末涂料中使用的光引发剂主要有自由基型和阳离子型。阳离子型光引发剂引发体系由于存在固化时间长、相对分子质量增长缓慢和少量的水对聚合反应有非常明显的阻聚作用等缺点,因此很难实现商业化和工业化。目前商业化应用比较广泛的还是自由基型光引发剂,它的优点是水对体系无阻聚作用、固化速率快,能够得到具有良好装饰效果和优良耐候性的涂层。
自由基型光引发剂有裂解型和提氢型两大类。提氢型引发剂主要有二苯甲酮类和硫杂蒽酮类等,这类 引发剂必须有供氢体 ( 胺、醇胺 ) 作为协同成分,否则无 引发效果。裂解型光引发剂多以芳基烷基酮衍生物为主,比较有代表性的有苯偶姻及其衍生物、苯偶酰缩酮 衍生物、二烷氧基苯乙酮、仅一羟烷基苯酮、仅一胺烷基 苯酮、酰基磷氧化物、芳基过氧化酯化合物、苯甲酰甲酸酯等。虽然目前市场上销售的光引发剂品种非常多,但却不适合制备较厚的固化材料,主要原因在于光引发剂的光敏感度和光引发活性还不够高;另外这些光引发剂品种都含有苯环或杂原子结构,由于含苯环或杂原子的自由基容易相互碰撞,产生带有发色基团的较大共轭体系,导致产品在使用过程中“黄变”。光引发剂的光敏感性不足和容易导致产品在使用过程中产生“黄变”这 2 个问题成为限制紫外光固化材料发展的瓶颈。因此设计开发含有多个乙酰基的新型高效光引发剂丙烯酸甲酯基三乙酰基甲烷,光敏感度和引发活性都将会非常地高,而且以乙酰基作为新型引发剂的自由基,不含苯环结构,难以生成较大共轭体系,可有效解决黄变问题。为了合成这个化合物,设计了 2 条合成路线。
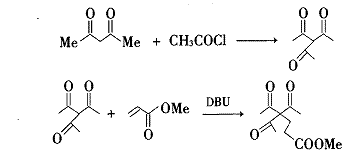
合成路线 I :
 合成路线Ⅱ
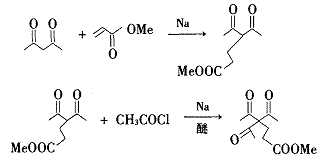
合成路线Ⅱ
 1 试验部分
1 . 1 仪器与试剂
1HNMR 、 13CNMR : Bluck400( 使用四甲基硅烷作标 准 ) ; GC : Agilent 6890 ; GC — MS : GC(6890N) , MS(Agilent 产的 5973N) 。
试剂和溶剂均为市售分析纯或化学纯。
1 . 2 试验步骤
1 . 2 . 1 合成路线 I
(1) 中间产物三乙酰基甲烷的合成
把 30 mL 乙醚加入到装有温度计、回流冷凝管、搅拌器的 100 mL 四口反应瓶中,接着加入 6 g (0 . 06mol) 乙酰丙酮,开始搅拌,使乙酰丙酮溶解到乙醚中。然后加入 0 . 69 g (0 . 03 mol) 金属钠。乙酰丙酮和金属钠反应结束并没有气泡放出后,滴加 2 . 355 g (0 . 03mol) 乙酰氯,然后加热回流,用 GC 检测反应过程。由于乙酰丙酮是酮式和烯醇式共存,而且与羰基相连的甲基上的氢原子也有一定的活性,因此反应非常复杂,目标产物三乙酰基甲烷收率非常低,仅为 30 %左右,而且比较难提纯。用同样的方法尝试了碳酸钾、叔丁醇钠、 DBU 、 NaH 等不同催化剂,但目标产物三乙酰基甲烷收率都非常低。
用以上碱性催化剂时,由于大部分乙酰丙酮是以烯醇式存在的,在乙酰丙酮的氧位置上易生成取代的副产物,因此想用 BF ,与乙酰丙酮的氧形成络合物,减少副产物,具体过程如下式:
1 试验部分
1 . 1 仪器与试剂
1HNMR 、 13CNMR : Bluck400( 使用四甲基硅烷作标 准 ) ; GC : Agilent 6890 ; GC — MS : GC(6890N) , MS(Agilent 产的 5973N) 。
试剂和溶剂均为市售分析纯或化学纯。
1 . 2 试验步骤
1 . 2 . 1 合成路线 I
(1) 中间产物三乙酰基甲烷的合成
把 30 mL 乙醚加入到装有温度计、回流冷凝管、搅拌器的 100 mL 四口反应瓶中,接着加入 6 g (0 . 06mol) 乙酰丙酮,开始搅拌,使乙酰丙酮溶解到乙醚中。然后加入 0 . 69 g (0 . 03 mol) 金属钠。乙酰丙酮和金属钠反应结束并没有气泡放出后,滴加 2 . 355 g (0 . 03mol) 乙酰氯,然后加热回流,用 GC 检测反应过程。由于乙酰丙酮是酮式和烯醇式共存,而且与羰基相连的甲基上的氢原子也有一定的活性,因此反应非常复杂,目标产物三乙酰基甲烷收率非常低,仅为 30 %左右,而且比较难提纯。用同样的方法尝试了碳酸钾、叔丁醇钠、 DBU 、 NaH 等不同催化剂,但目标产物三乙酰基甲烷收率都非常低。
用以上碱性催化剂时,由于大部分乙酰丙酮是以烯醇式存在的,在乙酰丙酮的氧位置上易生成取代的副产物,因此想用 BF ,与乙酰丙酮的氧形成络合物,减少副产物,具体过程如下式:
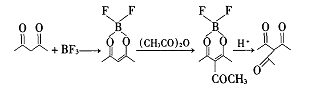 虽然该反应没有在氧位置上取代的副产物,但收率非常低,只有 18 . 2 %,原因可能是中间产物的络合能力太强,导致反应不易继续进行。
通过尝试以上合成中间产物三乙酰基甲烷的方法,用乙酰丙酮和乙酰氯为原料,以金属钠作催化剂的收率相对较高。用 1 mol 的乙酰丙酮做放大反应,得到三乙酰基甲烷,再进行下一步反应。
(2) 目标产物丙烯酸甲酯基三乙酰基甲烷的合成在装有温度计、回流冷凝管、搅拌器的 100 mL 四口反应瓶中加入 3 g (0 . 021 mo1) 三乙酰基甲烷和 30mL 干燥的 THF ,然后再缓慢加入 0 . 15 g (0 . 001 mol)DBU ,从温度计上观察到温度很快地上升,反应比较激烈。等温度无变化后,再把 1 . 81 g (0 . 021 mol) 丙烯酸甲酯滴加到反应瓶中。在加热回流条件下反应,用 GC 检测反应。
从 GC 图上看目标产物收率非常低,在反应 2 h 后补加 0 . 2 g 金属钠,收率有所提高,但最高只有 12 . 3 %。然后随着反应时间的延长,收率反而降低,估计是目标产物开始分解所导致。三乙酰基甲烷由于 3 个三乙酰基连在同一个碳原子上,也非常不稳定,受热容易分解。因此该反应路线不适合合成目标化合物。
1 . 2 . 2 合成路线 II
(1) 中间产物丙烯酸甲酯基乙酰丙酮的合成
将 80 g (0 . 8 mol) 乙酰丙酮加入到装有温度计、回流冷凝管、搅拌器的 500 mL 四口反应瓶中。接着加入 0 . 3 g (0 . 013 mo1) 金属钠,从温度计上观察到温度很快地上升,反应比较激烈。等温度冷却到室温后,把反应温度控制在 35 ℃ 以下,滴加 34 . 4 g (0 . 4 mol) 丙烯酸甲酯,滴加 1 h 后,将反应温度降至室温。
用 GC — MA 对单加成与双加成的产物进行确认,从 GC 图上看, 92 %是单加成产物。用盐酸把反应液的 pH 调节到中性,然后过滤掉盐,对反应液减压蒸馏,得到 72 g 纯度为 98 %的目标中间产物丙烯酸甲酯 基乙酰丙酮。
(2) 目标产物丙烯酸甲酯基三乙酰基甲烷的合成在装有温度计、回流冷凝管、搅拌器的 500 mL 四口反应瓶中加入 35 g (0 . 188 tool) 丙烯酸甲酯基乙酰丙酮和 200 mL 干燥的乙醚,然后再缓慢加入 4 . 33 g (0 . 188 mo1) 金属钠,从温度计上观察到温度很快地上升,反应比较剧烈。等温度无变化后,用 GC 检测不到丙烯酸甲酯基乙酰丙酮,说明金属钠已经与丙烯酸甲酯基乙酰丙酮反应完全。把温度降低到 20 ℃ ,再把蒸馏过的 14 . 76 g (0 . 188 mo1) 乙酰氯滴加到反应瓶中。在加热回流条件下反应 3 h ,用 GC 检测反应。 从 GC 图上看目标产物收率可达 88 . 3 % ,副产物比较少,比合成路线 I 效果要好很多。目标产物的收率比较高,通过减压蒸馏,可以得到纯度为 98 . 6 %的目标化合物 33 . 9 g ,总收率 85 %。该目标化合物的结构 经 1H — NMR 、 13C — NMR 和 GC — MS 表征如下:
虽然该反应没有在氧位置上取代的副产物,但收率非常低,只有 18 . 2 %,原因可能是中间产物的络合能力太强,导致反应不易继续进行。
通过尝试以上合成中间产物三乙酰基甲烷的方法,用乙酰丙酮和乙酰氯为原料,以金属钠作催化剂的收率相对较高。用 1 mol 的乙酰丙酮做放大反应,得到三乙酰基甲烷,再进行下一步反应。
(2) 目标产物丙烯酸甲酯基三乙酰基甲烷的合成在装有温度计、回流冷凝管、搅拌器的 100 mL 四口反应瓶中加入 3 g (0 . 021 mo1) 三乙酰基甲烷和 30mL 干燥的 THF ,然后再缓慢加入 0 . 15 g (0 . 001 mol)DBU ,从温度计上观察到温度很快地上升,反应比较激烈。等温度无变化后,再把 1 . 81 g (0 . 021 mol) 丙烯酸甲酯滴加到反应瓶中。在加热回流条件下反应,用 GC 检测反应。
从 GC 图上看目标产物收率非常低,在反应 2 h 后补加 0 . 2 g 金属钠,收率有所提高,但最高只有 12 . 3 %。然后随着反应时间的延长,收率反而降低,估计是目标产物开始分解所导致。三乙酰基甲烷由于 3 个三乙酰基连在同一个碳原子上,也非常不稳定,受热容易分解。因此该反应路线不适合合成目标化合物。
1 . 2 . 2 合成路线 II
(1) 中间产物丙烯酸甲酯基乙酰丙酮的合成
将 80 g (0 . 8 mol) 乙酰丙酮加入到装有温度计、回流冷凝管、搅拌器的 500 mL 四口反应瓶中。接着加入 0 . 3 g (0 . 013 mo1) 金属钠,从温度计上观察到温度很快地上升,反应比较激烈。等温度冷却到室温后,把反应温度控制在 35 ℃ 以下,滴加 34 . 4 g (0 . 4 mol) 丙烯酸甲酯,滴加 1 h 后,将反应温度降至室温。
用 GC — MA 对单加成与双加成的产物进行确认,从 GC 图上看, 92 %是单加成产物。用盐酸把反应液的 pH 调节到中性,然后过滤掉盐,对反应液减压蒸馏,得到 72 g 纯度为 98 %的目标中间产物丙烯酸甲酯 基乙酰丙酮。
(2) 目标产物丙烯酸甲酯基三乙酰基甲烷的合成在装有温度计、回流冷凝管、搅拌器的 500 mL 四口反应瓶中加入 35 g (0 . 188 tool) 丙烯酸甲酯基乙酰丙酮和 200 mL 干燥的乙醚,然后再缓慢加入 4 . 33 g (0 . 188 mo1) 金属钠,从温度计上观察到温度很快地上升,反应比较剧烈。等温度无变化后,用 GC 检测不到丙烯酸甲酯基乙酰丙酮,说明金属钠已经与丙烯酸甲酯基乙酰丙酮反应完全。把温度降低到 20 ℃ ,再把蒸馏过的 14 . 76 g (0 . 188 mo1) 乙酰氯滴加到反应瓶中。在加热回流条件下反应 3 h ,用 GC 检测反应。 从 GC 图上看目标产物收率可达 88 . 3 % ,副产物比较少,比合成路线 I 效果要好很多。目标产物的收率比较高,通过减压蒸馏,可以得到纯度为 98 . 6 %的目标化合物 33 . 9 g ,总收率 85 %。该目标化合物的结构 经 1H — NMR 、 13C — NMR 和 GC — MS 表征如下:
 2 结果与讨论
2 . 1 合成路线 I
合成路线 I 是先合成三乙酰基甲烷,然后再合成 目标产物丙烯酸甲酯基三乙酰基甲烷。但合成三乙酰 基甲烷的难度非常大,分别尝试了如试验部分所述的多种方法,结果都不理想,要么反应的收率非常低,要么反应非常难以进行。主要原因在于乙酰基是非常活泼的官能团, 3 个乙酰基连在同一个碳原子上,导致三乙酰基甲烷非常不稳定。在第 2 步反应:三乙酰基甲烷与丙烯酸甲酯的加成反应中,虽然能够得到目标产物,但收率非常低,原因在于乙酰基在碱性条件下非常容易脱去,这也证明了三乙酰基甲烷非常不稳定。
2 . 2 合成路线Ⅱ
合成路线Ⅱ是先合成丙烯酸甲酯基乙酰丙酮,然后再合成目标产物丙烯酸甲酯基三乙酰基甲烷。由于中间产物丙烯酸甲酯基乙酰丙酮比较稳定,因此合成的难度比较小;而且丙烯酸甲酯与乙酰丙酮的加成反应比较容易进行,试验证明金属钠作催化剂非常适合该类型的迈克尔加成反应,效率比较高。为了尽量减少双加成副产物,乙酰丙酮的量至少是丙烯酸甲酯的 2 倍,由于单加成与双加成产物的沸点差别较大,用减压蒸馏可把它们分开,从而得到高纯度的目标中间产 物丙烯酸甲酯基乙酰丙酮。
然后用中间产物丙烯酸甲酯基乙酰丙酮与乙酰氯反应合成目标产物,该反应也比较容易进行。用金属钠作催化剂和乙醚作溶剂效果比较好,目标产物的收率比较高,非常适合该类型的取代反应。通过减压蒸馏,可以得到高纯度的目标化合物,总收率 85 %。催化剂的碱性强度对该取代反应非常重要,丙烯酸甲酯基乙酰丙酮由于空间位阻增大,其 α — H 活性被大大降低,催化剂的碱性强度要足够高,才能提高反应收率。目标产物结构经 1H — NMR 、 13C — NMR 和 GC — MS 确认。
2 . 3 评价结果
该目标产物丙烯酸甲酯基甲烷放在 uV 固化粉末涂料中评价,有非常高的 uV 光引发效果,已经在中国、印度、日本、美国、德国、英国、法国等多个国家申请了专利。产品的工业化和商业化正在进行中。
2 结果与讨论
2 . 1 合成路线 I
合成路线 I 是先合成三乙酰基甲烷,然后再合成 目标产物丙烯酸甲酯基三乙酰基甲烷。但合成三乙酰 基甲烷的难度非常大,分别尝试了如试验部分所述的多种方法,结果都不理想,要么反应的收率非常低,要么反应非常难以进行。主要原因在于乙酰基是非常活泼的官能团, 3 个乙酰基连在同一个碳原子上,导致三乙酰基甲烷非常不稳定。在第 2 步反应:三乙酰基甲烷与丙烯酸甲酯的加成反应中,虽然能够得到目标产物,但收率非常低,原因在于乙酰基在碱性条件下非常容易脱去,这也证明了三乙酰基甲烷非常不稳定。
2 . 2 合成路线Ⅱ
合成路线Ⅱ是先合成丙烯酸甲酯基乙酰丙酮,然后再合成目标产物丙烯酸甲酯基三乙酰基甲烷。由于中间产物丙烯酸甲酯基乙酰丙酮比较稳定,因此合成的难度比较小;而且丙烯酸甲酯与乙酰丙酮的加成反应比较容易进行,试验证明金属钠作催化剂非常适合该类型的迈克尔加成反应,效率比较高。为了尽量减少双加成副产物,乙酰丙酮的量至少是丙烯酸甲酯的 2 倍,由于单加成与双加成产物的沸点差别较大,用减压蒸馏可把它们分开,从而得到高纯度的目标中间产 物丙烯酸甲酯基乙酰丙酮。
然后用中间产物丙烯酸甲酯基乙酰丙酮与乙酰氯反应合成目标产物,该反应也比较容易进行。用金属钠作催化剂和乙醚作溶剂效果比较好,目标产物的收率比较高,非常适合该类型的取代反应。通过减压蒸馏,可以得到高纯度的目标化合物,总收率 85 %。催化剂的碱性强度对该取代反应非常重要,丙烯酸甲酯基乙酰丙酮由于空间位阻增大,其 α — H 活性被大大降低,催化剂的碱性强度要足够高,才能提高反应收率。目标产物结构经 1H — NMR 、 13C — NMR 和 GC — MS 确认。
2 . 3 评价结果
该目标产物丙烯酸甲酯基甲烷放在 uV 固化粉末涂料中评价,有非常高的 uV 光引发效果,已经在中国、印度、日本、美国、德国、英国、法国等多个国家申请了专利。产品的工业化和商业化正在进行中。
合成路线Ⅱ
1 试验部分
1 . 1 仪器与试剂
1HNMR 、 13CNMR : Bluck400( 使用四甲基硅烷作标 准 ) ; GC : Agilent 6890 ; GC — MS : GC(6890N) , MS(Agilent 产的 5973N) 。
试剂和溶剂均为市售分析纯或化学纯。
1 . 2 试验步骤
1 . 2 . 1 合成路线 I
(1) 中间产物三乙酰基甲烷的合成
把 30 mL 乙醚加入到装有温度计、回流冷凝管、搅拌器的 100 mL 四口反应瓶中,接着加入 6 g (0 . 06mol) 乙酰丙酮,开始搅拌,使乙酰丙酮溶解到乙醚中。然后加入 0 . 69 g (0 . 03 mol) 金属钠。乙酰丙酮和金属钠反应结束并没有气泡放出后,滴加 2 . 355 g (0 . 03mol) 乙酰氯,然后加热回流,用 GC 检测反应过程。由于乙酰丙酮是酮式和烯醇式共存,而且与羰基相连的甲基上的氢原子也有一定的活性,因此反应非常复杂,目标产物三乙酰基甲烷收率非常低,仅为 30 %左右,而且比较难提纯。用同样的方法尝试了碳酸钾、叔丁醇钠、 DBU 、 NaH 等不同催化剂,但目标产物三乙酰基甲烷收率都非常低。
用以上碱性催化剂时,由于大部分乙酰丙酮是以烯醇式存在的,在乙酰丙酮的氧位置上易生成取代的副产物,因此想用 BF ,与乙酰丙酮的氧形成络合物,减少副产物,具体过程如下式:
虽然该反应没有在氧位置上取代的副产物,但收率非常低,只有 18 . 2 %,原因可能是中间产物的络合能力太强,导致反应不易继续进行。
通过尝试以上合成中间产物三乙酰基甲烷的方法,用乙酰丙酮和乙酰氯为原料,以金属钠作催化剂的收率相对较高。用 1 mol 的乙酰丙酮做放大反应,得到三乙酰基甲烷,再进行下一步反应。
(2) 目标产物丙烯酸甲酯基三乙酰基甲烷的合成在装有温度计、回流冷凝管、搅拌器的 100 mL 四口反应瓶中加入 3 g (0 . 021 mo1) 三乙酰基甲烷和 30mL 干燥的 THF ,然后再缓慢加入 0 . 15 g (0 . 001 mol)DBU ,从温度计上观察到温度很快地上升,反应比较激烈。等温度无变化后,再把 1 . 81 g (0 . 021 mol) 丙烯酸甲酯滴加到反应瓶中。在加热回流条件下反应,用 GC 检测反应。
从 GC 图上看目标产物收率非常低,在反应 2 h 后补加 0 . 2 g 金属钠,收率有所提高,但最高只有 12 . 3 %。然后随着反应时间的延长,收率反而降低,估计是目标产物开始分解所导致。三乙酰基甲烷由于 3 个三乙酰基连在同一个碳原子上,也非常不稳定,受热容易分解。因此该反应路线不适合合成目标化合物。
1 . 2 . 2 合成路线 II
(1) 中间产物丙烯酸甲酯基乙酰丙酮的合成
将 80 g (0 . 8 mol) 乙酰丙酮加入到装有温度计、回流冷凝管、搅拌器的 500 mL 四口反应瓶中。接着加入 0 . 3 g (0 . 013 mo1) 金属钠,从温度计上观察到温度很快地上升,反应比较激烈。等温度冷却到室温后,把反应温度控制在 35 ℃ 以下,滴加 34 . 4 g (0 . 4 mol) 丙烯酸甲酯,滴加 1 h 后,将反应温度降至室温。
用 GC — MA 对单加成与双加成的产物进行确认,从 GC 图上看, 92 %是单加成产物。用盐酸把反应液的 pH 调节到中性,然后过滤掉盐,对反应液减压蒸馏,得到 72 g 纯度为 98 %的目标中间产物丙烯酸甲酯 基乙酰丙酮。
(2) 目标产物丙烯酸甲酯基三乙酰基甲烷的合成在装有温度计、回流冷凝管、搅拌器的 500 mL 四口反应瓶中加入 35 g (0 . 188 tool) 丙烯酸甲酯基乙酰丙酮和 200 mL 干燥的乙醚,然后再缓慢加入 4 . 33 g (0 . 188 mo1) 金属钠,从温度计上观察到温度很快地上升,反应比较剧烈。等温度无变化后,用 GC 检测不到丙烯酸甲酯基乙酰丙酮,说明金属钠已经与丙烯酸甲酯基乙酰丙酮反应完全。把温度降低到 20 ℃ ,再把蒸馏过的 14 . 76 g (0 . 188 mo1) 乙酰氯滴加到反应瓶中。在加热回流条件下反应 3 h ,用 GC 检测反应。 从 GC 图上看目标产物收率可达 88 . 3 % ,副产物比较少,比合成路线 I 效果要好很多。目标产物的收率比较高,通过减压蒸馏,可以得到纯度为 98 . 6 %的目标化合物 33 . 9 g ,总收率 85 %。该目标化合物的结构 经 1H — NMR 、 13C — NMR 和 GC — MS 表征如下:
2 结果与讨论
2 . 1 合成路线 I
合成路线 I 是先合成三乙酰基甲烷,然后再合成 目标产物丙烯酸甲酯基三乙酰基甲烷。但合成三乙酰 基甲烷的难度非常大,分别尝试了如试验部分所述的多种方法,结果都不理想,要么反应的收率非常低,要么反应非常难以进行。主要原因在于乙酰基是非常活泼的官能团, 3 个乙酰基连在同一个碳原子上,导致三乙酰基甲烷非常不稳定。在第 2 步反应:三乙酰基甲烷与丙烯酸甲酯的加成反应中,虽然能够得到目标产物,但收率非常低,原因在于乙酰基在碱性条件下非常容易脱去,这也证明了三乙酰基甲烷非常不稳定。
2 . 2 合成路线Ⅱ
合成路线Ⅱ是先合成丙烯酸甲酯基乙酰丙酮,然后再合成目标产物丙烯酸甲酯基三乙酰基甲烷。由于中间产物丙烯酸甲酯基乙酰丙酮比较稳定,因此合成的难度比较小;而且丙烯酸甲酯与乙酰丙酮的加成反应比较容易进行,试验证明金属钠作催化剂非常适合该类型的迈克尔加成反应,效率比较高。为了尽量减少双加成副产物,乙酰丙酮的量至少是丙烯酸甲酯的 2 倍,由于单加成与双加成产物的沸点差别较大,用减压蒸馏可把它们分开,从而得到高纯度的目标中间产 物丙烯酸甲酯基乙酰丙酮。
然后用中间产物丙烯酸甲酯基乙酰丙酮与乙酰氯反应合成目标产物,该反应也比较容易进行。用金属钠作催化剂和乙醚作溶剂效果比较好,目标产物的收率比较高,非常适合该类型的取代反应。通过减压蒸馏,可以得到高纯度的目标化合物,总收率 85 %。催化剂的碱性强度对该取代反应非常重要,丙烯酸甲酯基乙酰丙酮由于空间位阻增大,其 α — H 活性被大大降低,催化剂的碱性强度要足够高,才能提高反应收率。目标产物结构经 1H — NMR 、 13C — NMR 和 GC — MS 确认。
2 . 3 评价结果
该目标产物丙烯酸甲酯基甲烷放在 uV 固化粉末涂料中评价,有非常高的 uV 光引发效果,已经在中国、印度、日本、美国、德国、英国、法国等多个国家申请了专利。产品的工业化和商业化正在进行中。
相关文章
发布评论
 已有
已有 杭州湖州绍兴马莱
杭州湖州绍兴马莱 巴洛克风格餐厅
巴洛克风格餐厅 马来漆工程097
马来漆工程097 马来漆工程095
马来漆工程095 马来漆工程094
马来漆工程094 马来漆工程086
马来漆工程086 马来漆工程082
马来漆工程082 马来漆工程078
马来漆工程078 马来漆工程069
马来漆工程069 马来漆工程065
马来漆工程065 马来漆工程061
马来漆工程061 马来漆工程060
马来漆工程060 马来漆工程051
马来漆工程051 马来漆工程036
马来漆工程036 马来漆工程034
马来漆工程034 马来漆工程031
马来漆工程031 马来漆工程028
马来漆工程028 马来漆工程001
马来漆工程001 马来漆工程002
马来漆工程002 马来漆工程003
马来漆工程003 马来漆工程004
马来漆工程004 马来漆工程005
马来漆工程005 马来漆工程006
马来漆工程006 马来漆工程007
马来漆工程007 马来漆工程008
马来漆工程008